Location: Home >> Detail
J Psychiatry Brain Sci. 2025;10(6):e250018. https://doi.org/10.20900/jpbs.20250018
 ,
Soheila Ansaripour 1 ,
Sadaf Eghtedari 1 ,
Zahra Heydari 1 ,
Vahid Farnia 2,3 ,
Mostafa Alikhani 2 ,
Safora Salemi 2 ,
Annette B. Brühl 4 ,
Dena Sadeghi-Bahmani 4,5 ,
Serge Brand 2,3,4,6,7,8,9,*
,
Soheila Ansaripour 1 ,
Sadaf Eghtedari 1 ,
Zahra Heydari 1 ,
Vahid Farnia 2,3 ,
Mostafa Alikhani 2 ,
Safora Salemi 2 ,
Annette B. Brühl 4 ,
Dena Sadeghi-Bahmani 4,5 ,
Serge Brand 2,3,4,6,7,8,9,*
1 Reproductive Biotechnology Research Center, Avicenna Research Institute (ARI), ACECR, Tehran 1941913114, Iran
2 Substance Abuse Prevention Research Center, Health Institute, Kermanshah University of Medical Sciences, Kermanshah 6719851151, Iran
3 Department of Psychiatry, School of Clinical Sciences, Faculty of Medicine, Nursing and Health Sciences, Monash University, Clayton, VIC 3168, Australia
4 Center for Affective, Stress and Sleep Disorders, Psychiatric Clinics, University of Basel, Basel 4002, Switzerland
5 Department of Psychology, Stanford University, Stanford, CA 94305, USA
6 Division of Sport Science and Psychosocial Health, Department of Sport, Exercise and Health, University of Basel, Basel 4052, Switzerland
7 Sleep Disorders Research Center, Department of Psychiatry, Kermanshah University of Medical Sciences (KUMS), Kermanshah 6714869914, Iran
8 School of Medicine, Tehran University of Medical Sciences, Tehran 1417466191, Iran
9 Center for Disaster Psychiatry and Disaster Psychology, Center of Competence of Disaster Medicine of the Swiss Armed Forces, Basel 4002, Switzerland
* Correspondence: Serge Brand
Background: About 4% to 12% of women in their fertile age suffer from polycystic ovarian syndrome (PCOS). Typically, women with PCOS report higher symptoms of depression, insomnia, and stress. To improve psychophysiological PCOS-related stress and insomnia, melatonin may have favorable effects. Accordingly, the present study aimed to investigate among women with PCOS, if melatonin improved symptoms of sleep disturbances and depression, compared to a placebo condition.
Methods: A total of 72 women (mean age: 31.21 years) with PCOS-related infertility were randomly assigned either to the melatonin or to the placebo condition. Melatonin/placebo intake lasted for six consecutive weeks (3 mg/d). At baseline and six weeks later at study end, participants completed a series of self-rating questionnaires on sleep patterns (Insomnia Severity Index (ISI); Pittsburgh Sleep Quality Index (PSQI); obstructive sleep apnea (OSA); restless legs syndrome (RLS)) and depression. After the study, participants underwent intracytoplasmic sperm injection (ICSI).
Results: OSA- and RLS-related values did not vary over time and within and between the melatonin and placebo conditions. Over time and irrespective of the melatonin or placebo condition, scores for insomnia decreased (significant p-value and medium effect size). Over time, scores for sleep disturbances descriptively decreased in the melatonin condition and descriptively increased in the placebo condition (medium effect size for the Time by Condition interaction). Next, scores for depression decreased descriptively over time, and irrespective of the study condition (always medium effect sizes).
Conclusions: The pattern of results suggests that, among women with PCOS and compared to placebo, overall, the effect of melatonin on sleep patterns and depression was modest. Future studies might focus on further standardized psychotherapeutic interventions, such as CBTi, to improve sleep and mood and to decrease PCOS-related psychosocial stress.
BDI-FS, Beck Depression Inventory-Fast Screen; CBTi, Cognitive Behavioral Therapy for Insomnia; ICSI, Intracytoplasmic Sperm Injection; ISI, Insomnia Severity Index; ITT, Intention-To-Treat; LH, Luteinizing Hormone; PCOS, polycystic ovarian syndrome; PSQI, Pittsburgh Sleep Quality Index; RLS, Restless Legs Syndrome
Polycystic ovary syndrome (PCOS) is one of the leading causes of anovulation and infertility and the most common metabolic disorder [1,2]. Prevalence rates vary between 4% to 12% in Europe [3]. Depending from the diagnostic criteria, more recent prevalence rates in Europe and the USA range from 6.2% to 19.5% [4], while the worldwide prevalence was 9.2% [1]. PCOS is commonly related to an androgen excess and ovarian dysfunction in the absence of other specific diagnoses [2]. At an observational level, women with PCOS report irregular menstruation, symptoms of hyperandrogenism (increased acne, seborrhea, hair loss on the scalp, increased body and facial hair), infrequent or absent menstruation [5], and infertility. Indeed, infertility in about 75% of women with PCOS is due to anovulation, making PCOS by far the most common cause of anovulatory infertility [6]. At a diagnostic level and following the Rotterdam criteria [7–9], the clinical diagnosis of PCOS requires two of the following three criteria: 1. Oligo ovulation or anovulation; 2. Clinical (including signs such as hirsutism) or biological (including a raised free androgen index or free testosterone) hyperandrogenism; 3. Polycystic ovaries are visible on ultrasound [2,7,8]. Further clinical manifestations may include menstrual irregularities, signs of androgen excess and obesity, along with insulin resistance, elevated serum LH, an increased risk of type 2 diabetes and cardiovascular events [2,7].
Compared to women with no PCOS, women with PCOS reported having lower quality of oocytes and embryos. To explain such observations, Azizi et al. [10] suggested the following underlying mechanisms: High insulin concentration (hyperinsulinemia) appears to induce androgens production and to reduce sex hormone-binding globulin (SHBG) synthesis; as a result, such endocrine dysfunctions result in hyperandrogenism [11]. As a further result, a high level of androgens both negatively impacts the communication between the oocyte and its companion granulosa cells and results in increased atretic and arrested follicles during the growing phase of PCOS ovaries [10,12]. Another explanation is the higher oxidative stress markers in the follicular fluid in women with PCOS, which may negatively impact infertility treatment outcomes. To counteract, melatonin has demonstrated antioxidant properties that protect oocytes from oxidative damage and may enhance oocyte quality and fertilization rates.
Thus, melatonin could be considered a potential adjunctive supplement for women with PCOS undergoing ICSI.
Further, a mental health-PCOS-link was observed: Compared to women with no PCOS, women with PCOS reported higher scores for mental disorders, in general [13–16], and for depression [17–26], anxiety [21,22,24,27], and for emotional distress [28], more specifically, along with a higher risk for self-harm [29].
Next, compared to women with no PCOS, women with PCOS reported higher scores for sleep disturbances [30–37], including OSA [34,38–42].
Overall, recent data suggest that compared to women without PCOS, women with PCOS report dramatically higher infertility rates and higher scores for sleep disturbances and depression.
The Associations between Melatonin, Ovarian Stimulation, Oxidative Stress, Sleep Regulation, and DepressionMelatonin is a monoamine neurotransmitter. In humans, melatonin is produced in the pineal gland functional already at birth [43] and exerts a broad variety of beneficial physiological modifications. It is commonly established that melatonin plays important roles in improving mitochondrial function [44–48]. Further, melatonin reduces the overall oxidative stress of the organism [49–57], and decreases insulin resistance [58].
Further, melatonin exerts a favorable effect on sleep patterns in the general population with sleep disorders [59–64], and above all, also among women with PCOS.
In line with this, there is also mounting evidence that exogenous melatonin administration exerts a favorable impact on mood, and above all on depression [65–69], and pain [70].
The Current StudyThere is sufficient research showing that compared to women without PCOS, women with PCOS suffer from higher scores for both sleep disturbances, including OSA and insomnia, and depression. By contrast, there is mounting evidence that melatonin has favorable effects on a broad variety of both physiological and psychological dimensions such as sleep and depression.
Although melatonin supplementation was initially considered for its potential fertility benefits, this study focuses on evaluating the psychological effects of melatonin in women with PCOS, scheduled to undergo ICSI, compared to a placebo.
Given this background, the following hypotheses were formulated.
First, following others [59–64], we hypothesized that compared to placebo, exogenous melatonin would improve sleep patterns, including OSA.
Second, based on previous results [65–69] we expected that compared to placebo, exogenous melatonin would improve symptoms of depression.
Between 2019 and 2020, we approached women with PCOS-related infertility with a subsequent plan of undergoing ICSI at the Avicenna Infertility Center (Tehran, Iran). Participants were fully informed about the aims of the study and the confidential and anonymized data handling. They were also informed that participation or non-participation had no impact on the current treatment. Thereafter, participants signed the written informed consent and completed a booklet of self-rating questionnaires covering sociodemographic information, sleep patterns, and depression (see details below). Next, participants were randomly assigned either to the melatonin or to the placebo condition. Both groups were assigned to standard treatment for ovarian stimulation for the subsequent ICSI. Further, at the end of the six-week-long study, participants completed again self-rating questionnaires.
ParticipantsInclusion criteria were: 1. Females aged between 20 and 40 years; 2. Diagnosis of PCOS based on the current Rotterdam criteria and ascertained by an experienced and clinically working gynecologist; 3. Planned to undergo ICSI for infertility treatment; 4. Signed written informed consent. Exclusion criteria were: 1. Withdrawing from the study; 2. Azoospermia in sperm analysis of male partner; 3. Further medical conditions such as an abnormal karyotype test, abnormal uterus anatomy, immune system or coagulation dysfunctions, always as ascertained by experienced medical doctors of the Avicenna Infertility Center (Tehran, Iran).
The flow chart (Figure 1) shows that out of the 100 individuals approached, 72 of those (72%) were randomized, and 65 (65%) completed the study. Data were analyzed per protocol.
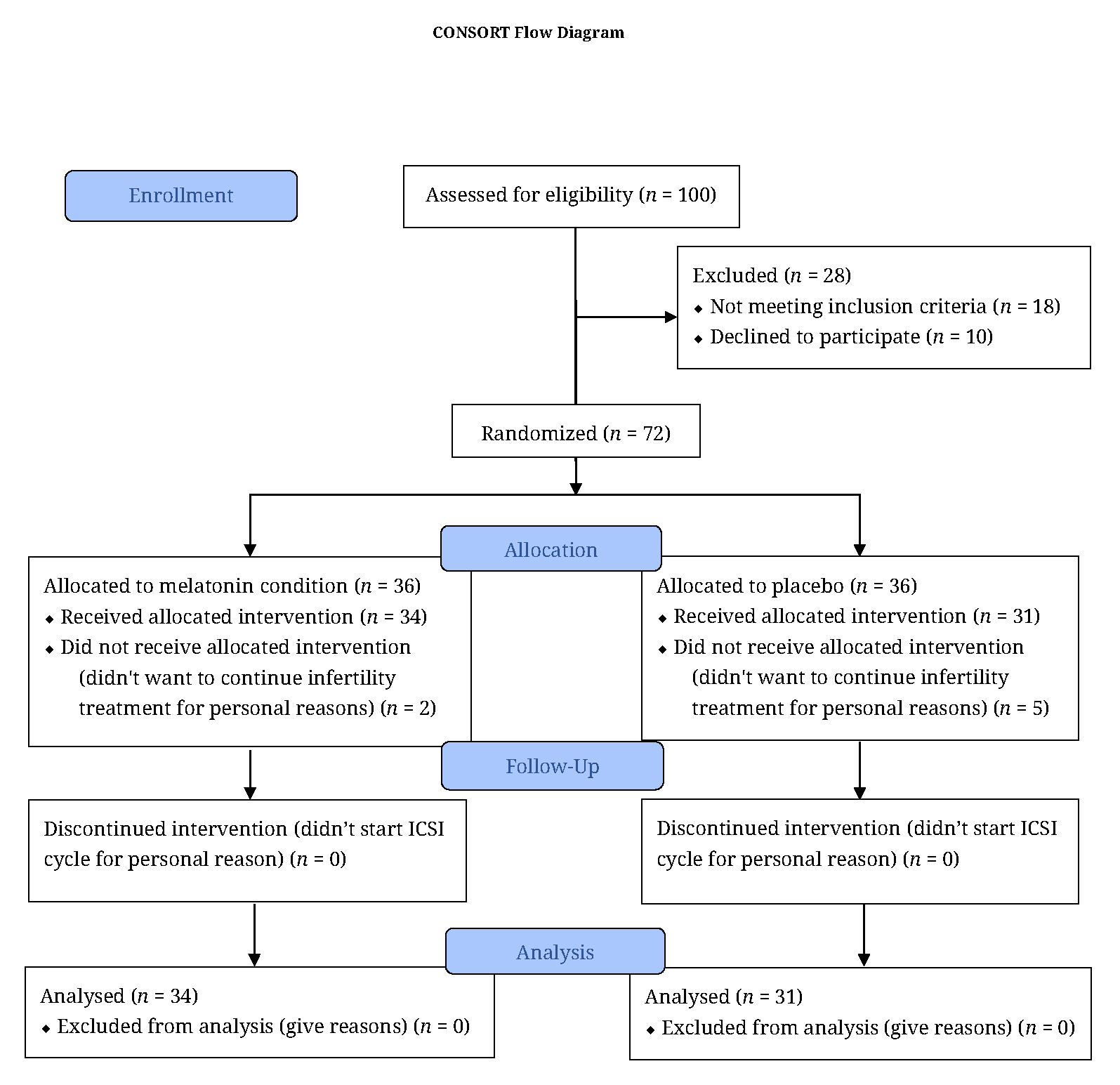 Figure 1. Patients approached, assessed, randomized, and statistically evaluated.
Figure 1. Patients approached, assessed, randomized, and statistically evaluated.
We used G*Power [71] to calculate the effect size. Following others [72] we expected a small effect size (f = 0.229; pη2: 0.050); alpha error: 0.05, power: 0.95; number of groups: 2; number of measurements: 2; total sample size: 64. We assessed 72 participants to counterbalance possible drop-outs.
RandomizationWe used a freely available randomization software (https://ctrandomization.cancer.gov) to randomly assign participants either to the melatonin or to the placebo condition. The software is a clinical trial randomization tool freely offered by the US American National Cancer Institute. A study nurse not further involved in the study cared for the randomization; participants and the handling medical doctors were blind to the participants’ assignment to the study condition.
General TreatmentIn both the melatonin and control conditions, following a three-week course of oral contraceptive pills (Ovocept-LD, Aburaihan Pharmaceutical Co., Tehran, Iran), Recombinant FSH (Cinnal-F, CinnaGen Co., Tehran, Iran) was started for ovarian stimulation on the third day of the subsequent menstrual cycle. The ovarian response was evaluated serially via transvaginal ultrasonography. When the leading follicles reached 12 mm in diameter, a GnRH antagonist (Cetrorelix 0.25 mg; Cetrotide, Merck Serono GmbH, Darmstadt, Germany) was introduced. Once the follicles reached an appropriate size (15–18 mm), final oocyte maturation was triggered using either a GnRH agonist (Triptorelin 0.1 mg; Variopeptyl, Varian Pharmed Co., Tehran, Iran) or HCG (5000 IU; Pregnyl, Merck Sharp & Dohme (MSD), Oss, The Netherlands). The oocyte retrieval was performed 36 h later under transvaginal ultrasound guidance and general anesthesia, followed by ICSI for fertilization.
Melatonin and PlaceboParticipants in the melatonin group received melatonin 3 mg tablets (Nature Made®, Pharmavite LLC, West Hills, CA, USA) daily in the evening, starting concurrently with the oral contraceptive pills and continuing until oocyte retrieval (a total duration of 6 weeks). The placebo group received identically appearing placebo tablets (matching in color, shape, size, and scent), also administered in the evening, starting concurrently with the oral contraceptive pills and continuing until oocyte retrieval (a total duration of 6 weeks).
Measures SleepPittsburgh Sleep Quality Index (PSQI)
Participants completed the Persian version [73,74] of the PSQI [75]. The PSQI is a self-rated questionnaire that takes approximately 5 minutes to complete. It consists of 19 items and contains seven subscales (subjective sleep quality, sleep latency, sleep duration, sleep efficiency, sleep disturbance, sleeping medication, daytime dysfunction); answers are given on 4-point Likert scales ranging from 0 (= not all all/never) to 3 (= always), with higher sum scores reflecting a poorer sleep quality (Cronbach’s alpha = 0.85).
Insomnia Severity Index (ISI)
Insomnia was assessed with the Persian version [76,77] of the ISI [78]. It consists of seven items, which refer in part to the Diagnostic and Statistical Manual of Mental Disorders [14,79] criteria for insomnia by assessing difficulty falling asleep, difficulty remaining asleep, early morning awakenings, impaired daytime performance, low satisfaction with sleep, and worry about sleep. Responses are recorded on a 5-point Likert scale ranging from 0 (= not at all) to 4 (= very much), with higher cumulative scores indicating greater severity of insomnia (Cronbach’s alpha = 0.89).
STOP-BANG Questionnaire
To self-assess symptoms of OSAs, participants completed the Persian version [80] of the STOP-BANG questionnaire [81]. It consists of four self-declared and OSAs-related items (STOP: Snoring, Tiredness, Observed apnea, and high blood pressure) and for demographic items (BANG: Body mass index [BMI; calculated as weight in kilograms divided by height in meters squared], Age, Neck circumference, and Gender) items. In the initial validation study, at a score of at least 3, the STOP-Bang questionnaire demonstrated a sensitivity of 84%, 93%, and 100% to detect all OSA (apnea-hypopnea index [AHI] ≥ 5), moderate OSA (AHI ≥ 15), and severe OSA (AHI ≥ 30), respectively [82].
Restless Legs Syndrome (RLS)
To assess RLS participants completed the Persian version [83] of the international RLS rating scale [84]. The self-rating questionnaire consists of ten items, and answers are given on 5-point Likert scales ranging from 0 (= never/not at all) to 4 (= always), with higher scores reflecting a higher RLS intensity.
Depression; Beck Depression Inventory-Fast Screen (BDI-FS)To assess symptoms of depression, participants completed the Persian version [85] of the BDI-FS [86]. It consists of seven items, and answers are given on 4-point Likert scales ranging from 0 (= no) to 3 (= a lot), with higher scores reflecting a greater severity of depressive symptoms (Cronbach’s alpha = 0.87).
Statistical AnalysisData were analyzed by protocol (melatonin condition: 36-2 = 34 full data set, placebo condition: 36-5 = 31 full data set). A provisional Intention-to-Treat (ITT) analysis did not yield another overall and significantly modified pattern of results.
To compare sociodemographic, PCOS- and ICSI-related information between the melatonin and placebo conditions, a series of X2- and t-tests was performed.
To compute whether dimensions of sleep and depression changed over time and between and within the melatonin and placebo conditions, a series of ANOVAs for repeated measures was performed with the following factors: Time (baseline; study end), Group (melatonin; placebo), and the Time x Group-interaction. Dependent variables were insomnia, sleep disturbances (PSQI), OSAs, RLS, and depression.
For F-tests, effect sizes are reported as partial eta squared (ηp2). The level of significance was set at alpha < 0.05. All statistical computations were performed with SPSS® 29.0 (IBM Corporation, Armonk NY, USA) for Apple Mac®.
Among the 72 participants initially assessed for the study, 34 participants in the melatonin group and 31 participants in control group continued the study (see flowchart).
General Demographic and Pregnancy-Related InformationTable 1 provides the descriptive and statistical indices of demographic and pregnancy-related dimensions.
No descriptively and statistically significant mean differences between participants in the melatonin and control condition were observed for age, BMI, duration of infertility, number of abortions, pregnancies, children, and the menstrual cycle pattern.
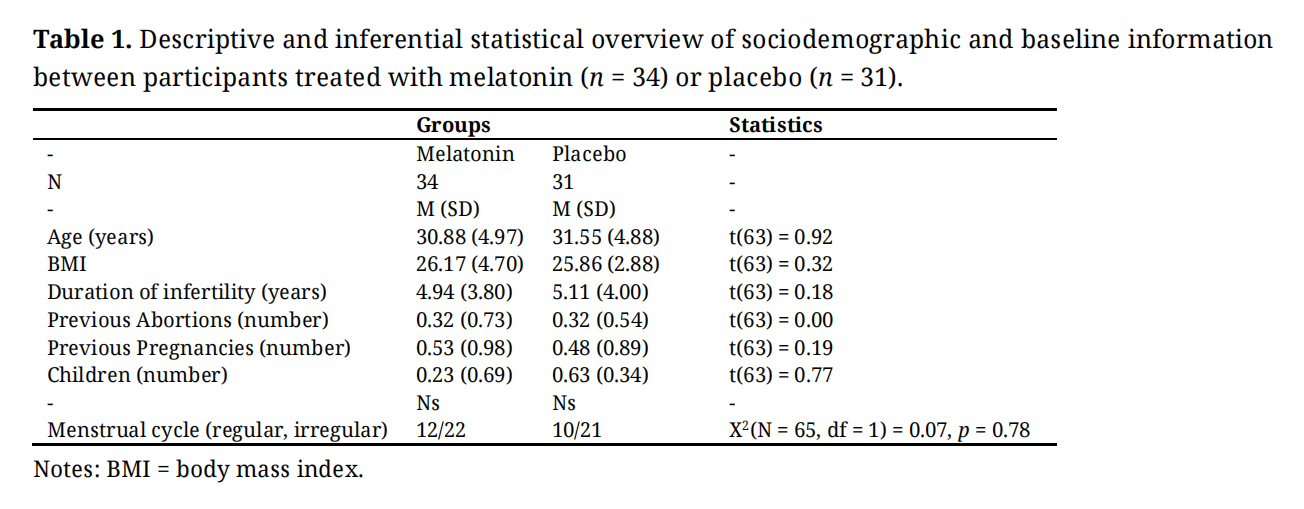 Table 1. Descriptive and inferential statistical overview of sociodemographic and baseline information between participants treated with melatonin (n = 34) or placebo (n = 31).
Table 1. Descriptive and inferential statistical overview of sociodemographic and baseline information between participants treated with melatonin (n = 34) or placebo (n = 31).
Table 2 provides the descriptive and statistical indices of the sleep-related dimensions and symptoms of depression.
OSA- and RLS-related values did not vary over time and within and between the melatonin and placebo condition. Over time and irrespective of the melatonin or placebo condition, scores for insomnia decreased (significant p-value and medium effect size). Over time, scores for sleep disturbances descriptively decreased in the melatonin condition and descriptively increased in the placebo condition (medium effect size for the Time by Condition interaction). Next, scores for depression decreased descriptively over time, and irrespective of the study condition (always medium effect sizes).
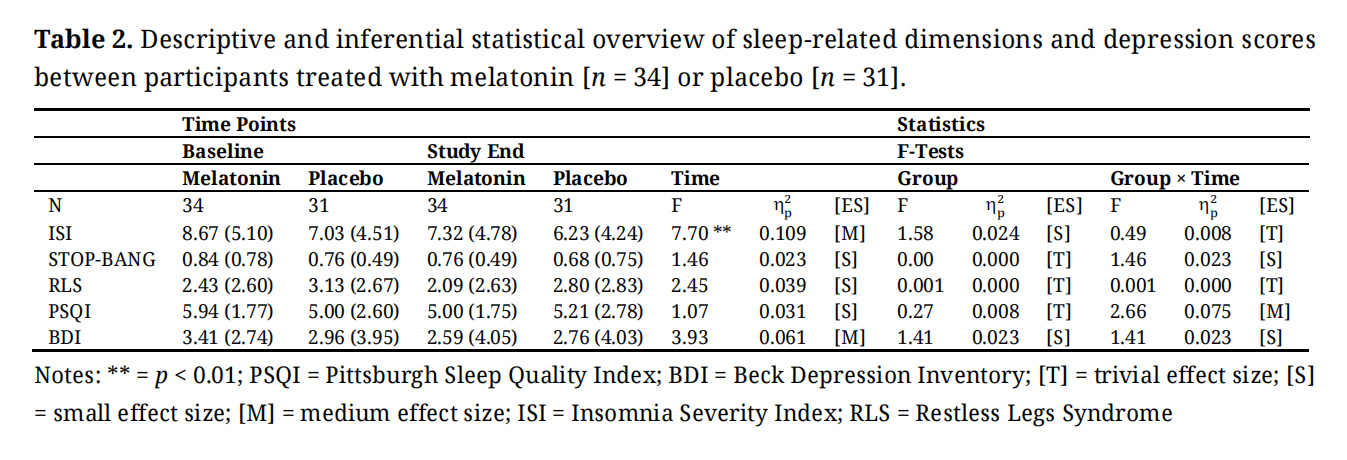 Table 2. Descriptive and inferential statistical overview of sleep-related dimensions and depression scores between participants treated with melatonin [n = 34] or placebo [n = 31].
Table 2. Descriptive and inferential statistical overview of sleep-related dimensions and depression scores between participants treated with melatonin [n = 34] or placebo [n = 31].
The aims of the present study were to investigate whether and to what extent compared to placebo, exogeneous melatonin administration might favorably impact sleep and depression among women with PCOS and undergoing subsequent ICSI treatment. Results showed that melatonin had no favorable (though also no unfavorable) effect on sleep and depression. A descriptive interaction effect in favor of melatonin was observed for one self-rating questionnaire assessing sleep disturbances (PSQI). The present results add to the current literature on women with PCOS and undergoing subsequent ICSI in the following two ways. First, against expectations, melatonin administration had no impact on sleep; likewise, second, melatonin administration had no favorable impact on depression. As such, to treat symptoms of sleep disturbances, including insomnia, OSA, and RSL, and depression among women with PCOS and undergoing subsequent ICSI, more effective treatments are needed.
Two hypotheses were formulated, and each of these is considered now in turn.
Melatonin and SleepWith the first hypothesis, we expected that compared to placebo, exogenous melatonin would improve sleep patterns, including OSA, and RLS, but data did not confirm this: Scores for insomnia improved over time irrespective of melatonin; scores for OSA, and RLS did not change, and the interaction effect (medium effect size) for the PSQI was due a descriptive worsening in the placebo condition, along with a descriptive improvement in the melatonin condition (see Table 2). As such, the present data are at odds with what has been observed before, both for the general population [59–64], and more specifically for women with PCOS [30–37], including OSA [34,38–42].
Though highly speculative, we suggest seven possibilities to explain the gap between the current literature and the present data. First, previous data were summarized in meta-analyses and systematic reviews among individuals with insomnia and general sleep complaints; as such, the comparison might be biased. Second, the comparison with women with PCOS might be biased, as women with PCOS in the present sample underwent subsequent ICSI treatment, which might be a further source of psychological distress. In fact, undergoing ICSI was associated with higher scores for psychological stress [87–90]. As such, future studies should consider psychological strain and stress as potential confounders. Third, the lack of significant results might be related to the measures themselves; indeed, while for the measures of insomnia (ISI), OSA (STOP BANG), and RLS no statistically and descriptively significant mean differences were observed, for sleep disturbances (PSQI), a nuanced improvement was observed with melatonin (medium effect size for the interaction; see Table 2). Given this, future studies might employ even more fine-grained measures to detect even more nuanced changes. Fourth, melatonin was simply not effective enough to improve sleep. Fifth, as shown in Table 2, participants were generally good sleepers (ISI cut-off values: ≤ 8 = no insomnia; ≤ 14 = subthreshold insomnia); as such, sixth, it appears unlikely that adjuvant melatonin could have had an adjunctive beneficial effect on participants’ good sleep. Seventh, the small statistical variances might have precluded yielding statistically significant mean differences between and within the study conditions and over time.
Melatonin and DepressionWith the second hypothesis, we expected that compared to placebo, exogenous melatonin would improve symptoms of depression, though, again, data did not support this assumption: Depression scores decreased over time irrespective of the study condition (see Table 2). Accordingly, results contrast with what has been observed elsewhere [65–69].
Again, the quality of the present data does not allow the explanation of the gap between the present pattern of results and previous studies. Tough, highly speculative, we advance the following five assumptions. First, given that women with PCOS might also suffer from a broad variety of further psychiatric issues, in general [13–16], including anxiety [21,22,24,27], emotional distress [28], and a higher risk for self-harm [29], it is conceivable that scores for anxiety, emotional distress, and self-harm have blurred the present pattern of results. Second, and relatedly, previous studies did not consider assessing women with PCOS undergoing subsequent ICSI, which might be a further source of psychological distress [87–90]. Third, similarly to the low sleep disturbance scores, participants had no mood-related issues (Table 2). As such, fourth, it appears unlikely that adjuvant melatonin could have had the power to exert an adjunctive beneficial effect on participants’ good mood. Fifth, the small statistical variances might have precluded to yield statistically significant mean differences between and within the study conditions and over time.
The strengths of the present study were the randomized double-blind study design, the precisely defined study population based on the Rotterdam criteria, the thorough assessment of sleep, including insomnia, sleep disturbances, including symptoms of OSA and RLS, the statistical approach focusing comprehensively on effect sizes, and not on mere p-values, and the study duration of six weeks, which should have allowed for observing improvements.
By contrast, limitations of the study were: 1. The small sample size, though, as mentioned above, we focused on effect size calculations, which are not sensitive to sample sizes. 2. As mentioned, latent and unassessed factors such as anxiety, undergoing ICSI, which per se might be a source of additional stress, emotional distress, or self-harm might have biased two or more dimensions in the same or opposite directions. 3. The measures might have been too coarse-grained to allow a more nuanced introspection of changes with melatonin. 4. We employed the standard medication of melatonin of 3 mg/d, though a higher dosage might have been more effective. 5. In this regard, a dose-response analysis might have further improved the overall pattern of results. 6. As already mentioned, participants had generally no sleep- and mood-related issues. 7. The different drop-out rates might have biased the overall pattern of results, though the ITT procedure, including the last-observation-carried-forward (LOCF), did not yield new findings or a modified pattern of results.
Future studies might enlarge the sample size, prolong the study duration to better capture potential effects of melatonin, including different melatonin dosages to get a better understanding of the melatonin-related dose-response relationship, assess more thoroughly PCOS- and ICSI-related stress, the degree of social support to counterbalance PCOS- and ICSI-related stress, and possible combinations of both melatonin and psychotherapeutic interventions such as specific Cognitive-Behavioral Therapy for insomnia (CBTi [91–94]) or Acceptance and Commitment Therapy [95,96].
Evidence from this randomized clinical trial study showed that oral melatonin supplementation had very modest effects on sleep and depression in women with PCOS undergoing subsequent ICSI.
All participants gave their signed written informed consent letters. Iranian Registry of clinical trials (Tehran, Iran) (IRCT20160717028967N10) and Ethical Committee of Avicenna Infertility Treatment Center (Tehran, Iran) approved the study (reference number; IR.ACECR.AVICENNA.REC.1398.002). The study performed under ethical principles contained in the 7th and current (2013) editions of Helsinki Declaration [97].
Declaration of Helsinki STROBE Reporting GuidelineThis study adhered to the Helsinki Declaration. The Strengthening the Reporting of Observational studies in Epidemiology (STROBE) reporting guideline was followed.
Data may be made available under the following conditions: 1. An expert in the field asks for data. 2. There must be robust and strong hypotheses to support and justify the request. 3. A statement and proves are needed to make sure that data are securely stored. 4. A statement is needed to make sure that data are not shared with third parties.
KY and SA conceived, designed, evaluated and drafted the manuscript; SE and ZH contributed to designing evaluation, collecting data and drafting the manuscript; SB, ABB, DS-B, and VF re-evaluated data, performed statistical analyses, interpreted findings and revised the manuscript; KY, VF, SB, ABB, DS-B, SS and MA interpreted findings and revised the manuscript; all authors read and approved the final manuscript.
Authors declare that they have no conflicts of interest.
This paper is the result of findings of research project No. 970010-048 approved and funded by Reproductive Biotechnology Research Center, Avicenna Research Institute (ARI), ACECR, Tehran, Iran.
Authors would like to thank the Reproductive Biotechnology Research Center, Avicenna Research Institute (ARI), ACECR, Tehran, Iran, for their support, cooperation and assistance throughout the study.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
Yazdchi K, Ansaripour S, Eghtedari S, Heydari Z, Farnia V, Alikhani M, et al. In women with polycystic ovarian syndrome (PCOS), oral melatonin supplementation had a modest effect on sleep and no effect on depression—results from a randomized clinical trial. J Psychiatry Brain Sci. 2025;10(6):e250018. https://doi.org/10.20900/jpbs.20250018.

Copyright © Hapres Co., Ltd. Privacy Policy | Terms and Conditions




