Location: Home >> Detail
J Psychiatry Brain Sci. 2018; 3(6): 11; https://doi.org/10.20900/jpbs.20180011

Paul Chook Department of Information Systems & Statistics, CUNY/Baruch College, 1 Bernard Baruch Way, New York, NY 10010, USA.
This article belongs to the Virtual Special Issue "Deep Phenotyping of Psychiatric Diseases"
Be careful what you wish for, lest it come true.
W.W. Jacobs, “The Monkey’s Paw”
In the past three decades, advances in molecular biology and high throughput platforms have led to an unprecedented number of novel findings about genomes, transcriptomes and proteomes. Indeed, we are now in the midst of what researchers have described as the “-omics” era. The voluminous aggregate of information across various -omic levels has created an ever-increasing number of opportunities to establish genotype-phenotype associations.
The many discoveries of multiple pathways to diseases and disorders from their inferred genetic origins are nothing short of breath-taking. The seminal concept of the genotype first presented by Johannsen [1], the searches for which many decades later would lead to studies of linkage disequilibrium, and from there to GWAS studies of SNPs, is lucidly summarized in Altshuler et al. [2]. In its ability to sequence many small pieces of DNA in parallel, Next Generation Sequencing (NGS), described as the most important tool in medicine since X-rays were invented [3], is now the preferred approach by which to examine gene expression [4]. In tandem with bioinformatics algorithms and protocols to assemble the genetic pieces into a complete genome, NGS can now be employed to rapidly detect a wide array of genetic abnormalities [5].
Many researchers now maintain that the next step in establishing fruitful genotype-phenotype relationships between genetic mutations and diseases/disorders will be to develop more thorough descriptions of the phenotype—Next Generation Phenotyping [3] or “deep phenotyping” [6]. In a somewhat related article, Joober [7] asserts that the most important objective of the 1,000 Genomes Project is identify the greatest number of rare genetic variants as they relate to neuropsychiatric disorders. This would be achieved by selecting a large, representative sample of the general population, then partitioning the sample into two components: those with rare variants and those without. Given the presumed prevalence of rare genetic variants along with other, statistical conjectures, Joober [7] asserted that 45,000 participants randomly sampled from the population would suffice, assuming that researchers could obtain deep phenotypes of all study subjects.
Similarly, Haring and Wallaschofski [8] proposed a “systems epidemiology” approach that would integrate several levels of -omic databases with additional environmental data to construct a multi-dimensional network model to aid in predicting causes of health and disease. According to these authors, in her the protocols and algorithms used to establish genotype-phenotype paradigms are robust.
Geschwind and Konopka [4] also suggest a systems level approach in neuroscience akin to the model presented by Haring and Wallaschofski [8]. However, Geschwind and Konopka [4] advocate the use of -omics as an exploratory, not confirmatory, procedure by which to ascertain genotype-phenotype associations related to neuropsychiatric and other brain-related disorders.
“When I make a word do a lot of work like that,' said Humpty Dumpty, 'I always pay it extra.”
—Lewis Carroll, the author of “Through the Looking Glass”.
To analyze and incorporate the expression deep phenotyping into the genotype-phenotype paradigm, a brief account of the origin and development of the concepts will, hopefully, provide a useful perspective. In 1911, Johannsen proposed the concepts of gene, genotype, and phenotype. The genotype was “the sum total of all genes”, and the phenotype evidenced by “direct inspection… or finer measures of assessment” of the organism under consideration. Embedded in Johannsen’s conception of the genotype-phenotype relationship was a model of causality: the genotype of the parent transmitted to the offspring produced the latter’s phenotype [9].
Concepts of clinical taxonomies—phenotypes—were developed much earlier, during the 17th and 18th centuries. The first recorded medical/clinical nosological systems were developed by Thomas Sydenham [10] and François Bossier de Sauvages de Lacroix [11]. The earliest systematic attempt to identify causal relationships in psychiatric disorders can be traced to the vicar Robert Burton, who published his Anatomy of Melancholy in 1621. Burton cataloged an assortment of causes of what we would now refer to as depression, citing “a heap of accidents” as one in particular.
Fast forward to the late 20th century and to the International Statistical Classification of Diseases and Related Health Problems (ICD-10) [12], a 3-volume tome containing 8,000 categories and 3-digit alphanumeric category codes; and, to the 21st century, in the United States, the Diagnostic and Statistical Manual of Mental Disorders-V (DSM-V) [13], which contains the most recent updating efforts by the American Psychiatric Association to standardize and classify all mental disorders. While there remain some questionable features of ICD-10 and DSM-V taxonomies, their attempts to standardize and classify diseases/disorders as completely and extensively as possible are much expanded and developed versions from earlier depictions of the medical phenotypes contained therein.
Other attempts to systematize and refine nomenclature in medical genetics were developed by John Carey and colleagues and known as the Elements of Morphology project. The impetus for the project was the apparent need to find a common language for phenotypes as they came to be associated with newly discovered genes. The objective was to standardize terminology in clinical morphology and syndromology [14]. Their efforts resulted in the publication of a series of six articles targeting specific features of the human clinical phenotype [15–20]. With its highly detailed descriptions and metrics, Carey and his colleagues produced the antecedents of what may now be referred to as deep phenotyping. A recent update referring to deep phenotyping is provided by Carey [21].
At about the same time, Cotton et al. [22] proposed the creation of the Human Variome Project (HVP). The HVP was a more wide-ranging approach that would examine comprehensive and accessible databases of neurogenetic disorders as well as incorporate genetic information from GWAS studies. Similar to the efforts by Carey et al. [14], the aims of the HVP were to adopt standards and develop algorithms for assessing the pathogenicity of neurogenetic disorders.
Coincident with these approaches, Altshuler et al. [2] also suggested that subjects of interest be “deeply phenotyped” in order to establish valid genotype-phenotype relationships. A year earlier, Tracy [6] contended that understanding genotype-phenotype relationships could be improved by deep phenotyping; that a fine-grained depiction of the phenotype would act to illuminate the pathways from genotype to phenotype. In 2012, a special issue of articles related to deep phenotyping as a means by which to attain a more precise and comprehensive assessment of diseases/disorders was published in Human Mutation. Robinson [23] contended that, in order for clinicians to make accurate diagnoses—“precision medicine”—a complete and comprehensive understanding of the features of any disease/disorder—a deep phenotype—was essential. Robinson [23] cited the Human Phenotype Ontology (HPO) group project (updated in Kohler et al. [24], which had collected more than 10,000 terms by which abnormal phenotypes could be described, that was superior to earlier bioinformatics tools [25], and could be used to link a given phenotype to disease genes [26,27].
Another effort to implement the use of deep phenotyping has been to standardize and utilize Electronic Health Records (EHR) to make available the mountains of clinical data stored therein to further elaborate and refine understanding of the causal mechanisms of diseases/disorders. Frey et al. [28] suggest that, by combining genomic data with deep phenotyping, bioinformatics algorithms could then identify, extract and cluster together the many salient features of the disease/disorder phenotype from EHRs and assess the diagnostic accuracy—its sensitivity and specificity—of the genetic abnormalities. Frey et al. [28] also proposed that the HPO terminology database be employed to identify categories of disease/disorder phenotypes reliably.
Longitudinal clinical deep phenotyping is purported to have been the source of several successes. For example, Chawes et al. [29] were able to identify cord blood Vitamin D deficiency in pregnant mothers as a causal factor of asthma- and allergy-related illnesses in pre-school age offspring. Deep phenotyping has also been alluded to as the best method by which to provide genetic counseling to women impregnated by In Vitro Fertilization (IVF) as to the most likely successful outcome of the procedure [30]. Results from their Receiver Operating Curve (ROC) analysis of the data showed that the 52-variable model employed by these researchers produced greater accuracy as measured by the Area Under the Curve (AUC) compared to the older, Templeton model (80% vs. 68%). In studies using mouse models of Huntington’s disease, Alexandrov et al. [31] found that by deeply phenotyping complex changes in behavior, they were able to predict more accurately the relationship between Cytosine-Adenine-Guanine (CAG) length and age of onset than did the simpler linear model of Age of onset regressed on CAG length alone. Using their proprietary software and machine-learning algorithms, Ruderfer and Dudley [32] found a set of approximately 200 behavioral features which accurately predicted the length of CAG repeats in strains of mice with the huntingtin gene.
“Human beings are complex and dangerous creatures.”
—Andre Gregory, the actor in “My Dinner with Andre”
Given the enormous complexity of the genome and phenotype, Altshuler et al. [2] cautioned that there may be built-in limitations to researchers’ abilities to establish valid genotype-phenotype relationships because of the need to examine extremely large samples of disease and non-disease cohorts. Sewell Wright [33] first acknowledged the problem where there exists a “complex of interacting, uncontrollable, and often obscure causes”. Similarly, the number of opportunities big data has for making errors increases markedly as the size of the database increases. Kohane et al. [34] referred to such events as the “incidentalome”, which threatens to undermine the -omic revolution.
As -omic databases become increasingly elaborate, massive and multiplex, and a growing number of common mutations—insertions, deletions, inversions, duplications—and variants in general found, questions arise as to which of the many common variants may have been identified correctly as the causal mechanisms for a disease/disorder, how many might have been falsely identified, and how many might have been missed. As an example, Kohane et al. [35] cited the publication of James Watson’s genome, which contains variants known for producing congenital disorders, none of which were diagnosed for Watson.
False identification problems associated with multiple comparisons identifying statistically significant associations of specific genotypes with diseases/disorders has been known for decades (See, for example, Austin et al. [36]). Solutions for controlling experimentwise Type I errors or false discovery rates (FDRs) in genomic and other big data sets range from the conservative procedures established by Bonferroni and Sidak et al. [37] to the more recently developed adjustments of FDRs [38–40] and posterior error probabilities [41]. Unfortunately, the more conservative procedures incur a substantial loss of power, while FDR methods are perceived as a tolerable compromise between inflated, unadjusted Type I experimentwise errors and the conservative, e.g., Bonferroni approach [39]. It should also be noted that some FDR methods for simulating null distributions are more limited than others in correctly identifying genes correlated with a phenotype [42]. Whatever method is employed, the most reliable course to follow has always been replication [43]. Unfortunately, this would involve additional considerable cost and time needed to recruit participants, and would not likely be fundable.
Recent studies of the actual levels of FDRs have raised additional concerns. Jager and Leek [44] examined p-values from five prestigious medical journals dating from 2000 to 2010 and found the “science-wise” rate of FDRs was 14%, a pronounced increase above the anticipated 5% level. Nonetheless, other discussants of the article by Jager and Leek [44] examined their data and found substantially higher rates. By raising the p-value threshold a trifle from p < 0.05 to p ≤ 0.05, Benjamini and Hechtlinger [45] estimated the science-wise FDR at 20.5%. Ioannidis [46] noted many other concerns related to the results obtained by Jager and Leek [44]; specifically, the variability of false positive (FP) rates across research areas and among study designs, a failure to acknowledge bias in the publications examined, bias in the selection of p-values from abstracts, and bias resulting from the highly select nature of the journals chosen for the sample. As Benjamini and Hechtlinger [45] remarked, “Modern science faces the problem of selection of promising findings from the noisy estimates of many” [45]. What is more, while these authors also note that these issues are often addressed in studies of genomics, proteomics and neuroimaging, the hazards associated with examining many thousands of parameters requires further, continual vigilance.
Despite its many strengths as a prototypical deep phenotype database, the HPO terminology website [47] contains a number of omissions and misstatements that could affect an accurate determination of a genotype-phenotype linkage, especially terms associated with neuropsychiatric and brain-related disorders. For example, a search of the term, “intellectual disability” (ID) produced the following: “Subnormal intellectual functioning which originates during the developmental period… defined as an IQ score below 70”. This is only partly correct. ID may appear (DSM says “occur”) during post-natal development, but more likely have its etiology in genetics or prenatal neurodevelopment. According to DSM, the developmental period extends from childhood to adolescence, after which such deficits should be diagnosed as a “neurocognitive disorder”. The definition of intellectual disability is also more elaborate, and requires the child to have “deficits or impairments in adaptive behavior” as measured by “standardized, culturally appropriate tests”. In the Synonyms section, the term, “mental handicap”, an expression often used by European pediatricians and child psychiatrists, is not included. A search of the term Learning Disability brings the reader to Specific Learning Disability. Under Synonyms, no terms are found. Yet, learning disability is often referred to as borderline ID, a term not listed in the HPO. Many genotypes for ID are listed, but Jacobsen Syndrome, generated by deletion 11q24–25, which produces both borderline disability and ID, is not listed as a possible genetic cause.
Clinical studies making use of deep phenotyping may undergo other, methodological challenges. For example, the study by Chawes et al. [29], recruited only mothers diagnosed with asthma. As Holland [48] once remarked, it takes two causes to define an effect: the treatment and the control. A prospective cohort study that included a control group of Vitamin-D deficient mothers without asthma would have reduced the possible bias in the results. The IVF study by Banerjee et al. [30] used a cross-validation procedure to identify significant predictor variables from the 52-variable set. However, these researchers found that 21 of the 52 variables in the training set were significantly different from those found in the validation data set, indicating that the factors predicting sources of variation in IVF outcome produced two markedly different models for the training and validation sets. Also, there was no indication as to whether the AUC from the Banerjee model (80%) was statistically significantly different from the Templeton model (68%), or if there was no overlap in the confidence intervals of the AUC percent estimates between the two models. In their study using a mouse model to examine deep phenotyping in Huntington’s disease, Ruderfer and Dudley [32] identified a set of approximately 200 behavioral variables to accurately predict CAG length. Having measured 3,086 behavioral features in mice with the Huntington mutation, Alexandrov et al. [31] found they required more than 200 variables to predict more accurately (R2 = 90%) CAG length. However, in an earlier, large multicenter study of 3,452 human participants, Langbehn et al. [49] found their simpler logistic regression model—as opposed to the traditional proportional hazards model—very accurately predicted age of onset of Huntington’s disease, using only CAG repeats as the predictor variable.
Prediction accuracy of diagnostic assessment often uses the ROC by constructing a scatterplot of the sensitivity [true positive (TP) rate] and false positive (FP) rate. When TP = FP, assignment to either category is equally likely, and the AUC is 50%. When TP is much greater than FP, the area under the curve becomes increasingly large, approaching 100%—the diagnostic instrument is highly sensitive and specific in detecting an abnormality. In their study relating gene expression to phenotypic data, Feiglin et al. [50] integrated 6,665 transcriptomes derived from tissues with 3,397 diseases. These investigators found only relatively weak genotype-phenotype associations between levels of expression and phenotype, with a maximum AUC = 0.69. Differences between patients with Alzheimer’s disease and non-disease controls based their respective gene expression for a specific SNP type as a basis for future diagnosis were described by Geschwind and Konopka [4]. In their Fig. 1 [4] in which genetic polymorphism was correlated with gene expression, there is a large overlap—false positive and false negative—in the gene expression variability between SNP types for patients with Alzheimer’s disease compared to controls.
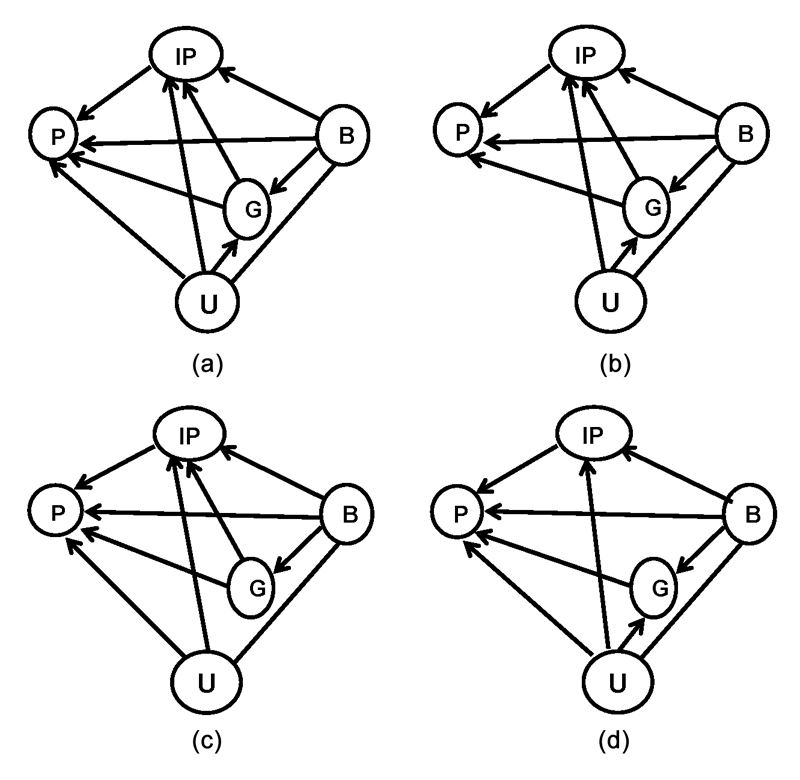 Fig. 1 Four examples of complex genotype-phenotype pathways: (a) the Phenotype (P) is contingent upon Genetic (G) factors, Intermediate Phenotype (IP) factors, Background (B) factors, and Unknown (U) factors. In addition, G and IP factors are contingent upon B and U, and, G is contingent upon U and B. (b) The same model as in (a), except P is not contingent upon U. (c) The same model as in (a), except G is not contingent upon U. (d) The same as in (a), except (IP) is not contingent upon (G).
Fig. 1 Four examples of complex genotype-phenotype pathways: (a) the Phenotype (P) is contingent upon Genetic (G) factors, Intermediate Phenotype (IP) factors, Background (B) factors, and Unknown (U) factors. In addition, G and IP factors are contingent upon B and U, and, G is contingent upon U and B. (b) The same model as in (a), except P is not contingent upon U. (c) The same model as in (a), except G is not contingent upon U. (d) The same as in (a), except (IP) is not contingent upon (G).
Clustering procedures employed by algorithms and based on “meaningfulness” of terms can be problematic, as Google and Facebook have discovered to their dismay. In their study of deep phenotyping serious mental illness, Jackson et al. [51] found that clinicians often used the same terms in different ways, and that clustering techniques used to identify meaningfulness may not establish reliable diagnoses. Using SNOMED CT mapping (an organized system of medical and related terms for clinical documentation and reporting), 557 curated concepts by 2 expert psychiatrists produced a probability-adjusted kappa coefficient of agreement of қ = 0.45, which is relatively modest.
Integrating EHR deep phenotyping with -omic data also presents challenges. Some information written into EHRs is incorrect. Clinical terms reported at different points in time or in different epochs may not have the same connotation, but may result in being clustered together. However, Son et al. [52] used EHR-Phenolyzer, an algorithm for extracting and analyzing phenotypes from heterogeneous EHR narratives. Results from their phenotypic analyses identified genes associated with confirmed monogenic disorders in 16/28 patients assessed.
Sometimes information is missing. Filtering EHRs containing large numbers of quantitative and qualitative information will also prove demanding. According to Hripcsak and Albers [53], Columbia University’s database contains 136,035 different concepts. As Hripcsak and Albers [53] observe, data may be biased, particularly when dealing with neuropsychiatric or neurodevelopmental disorders. The CDC computes its prevalence of autism spectrum disorders (ASD) based on medical, clinical, and school records. However, there is much anecdotal evidence to suggest that pediatricians, child psychiatrists and psychologists, as well as nurses and other clinicians, have been pressured by parents to assign a diagnosis of ASD rather than ID or learning disability, and for various reasons. Such recorded information would erroneously inflate the prevalence of ASD in the population, not to mention undermine attempts to ascertain valid genotype-phenotype relationships.
As this latter example suggests, developing a proper taxonomic system of neuropsychiatric and neurobehavioral disorders can be quite demanding and controversial, and has been for many years. In order to identify their genetic origins, Kendler [54,55] has argued that psychiatric nosology be systematized in such a way that the diagnostic criteria used to classify disorders produce both high sensitivity (TP) and high specificity (TN). To that end, the DSM manual continually updates the set of criteria it uses to increase diagnostic accuracy for specific psychiatric disorders. Unfortunately, DSM revisions have had unintended consequences. For example, ASD as currently defined was originally classified as Childhood Schizophrenia, the latter diagnosis of which persisted until DSM-III was published in 1980. In addition, one of the criteria—age of onset—has been modified several times in the past 40 years. In DSM-III [56] age of onset was set as prior to 30 months; in DSM-IIIR [57], it was increased to 36 months; in its most recent incarnation, “individuals with ASD must show symptoms from early childhood, even if those symptoms are not recognized until later” [13]. Moreover, over the last 50 years, each time DSM criteria for ASD have been revised, the prevalence of the disorder shifts noticeably upward [58].
Additional and often neglected sets of factors in genotype-phenotype models affecting diseases/disorders are stochastic factors related to human development, and to exogenous environmental factors. A meta-analysis of twin studies of diseases/disorders over the past half century has again shown that genetic contributions to psychiatric and behavioral disorders have a high heritability rate, based on strong correlations of disorders among monozygotic twins [59]. However, high heritability may not translate into consistent genetic models of causality [60,61]. The complexity of the phenotype likely increases with the individual’s age as a result of the extent to which other genes have had time to interact with the genetic cause. In that regard, Polderman et al. [59] found that correlations decrease as twins age, suggesting that there are important developmental and/or environmental components modifying many neuropsychiatric and behavioral disorders. It should be noted that, in their study of deep phenotyping, Stepniak et al. [62] found that environmental but not genetic factors were significantly associated with early onset schizophrenia.
These last remarks provide additional methodological complications that will influence how deep phenotyping of neuropsychiatric disorders can be used successfully to ascertain genetic causes. As a consequence, current models that have applied various -omic levels with deep phenotyping may well be incomplete. In discussing features of his path models, Wright [33] was prescient in anticipating that there may be multiple paths other than the known or suspected factors producing the phenotype.
One approach that had been proposed to identify novel genotype-phenotype pathways was the concept of the endophenotype proposed by Gottesman and Shields [63]. These authors suggested that a well-defined and quantifiable intermediate phenotype could be used to elucidate the causal path from genetic sources to the phenotype, schizophrenia. In this -omic era, strata containing transcriptomes and/or proteomes could be considered as intermediate phenotypes. However, as noted earlier, introducing intermediate phenotypes into causal modeling will invariably complicate the process of identifying authentic genotype-phenotype pathways. Moreover, as Cox and Wermuth [64] note, the presence of unidentified “lurking” variables in observational studies, the absence of which may inadvertently affect the interpretation of observed relationships, will have an adverse effect on the causal model constructed.
The causal models in Fig. 1 illustrate four possible complications that may arise in constructing a causal model when there are unidentified confounding factors. In Fig. 1(a), the unidentified variables (U) are contained within the background factors (B), but also directly affect the phenotype (P), the intermediate phenotype (IP), and genotype (G). Fig. 1(b) illustrates the same relationships of U with IP and G, but not P. Fig. 1(c) exemplifies a causal model in which U influences P and IP, but is unrelated to G. The fourth figure characterizes a causal model in which U influences G, IP, P, but the genetic factor, G, is not.
While it is clear that the -omics era has introduced many new technologies across several levels of biological strata that could eventually lead to important findings about the complex nature of the genotype and its various intermediate phenotypes, it is as important to remember that by incorporating deep phenotyping into the genotype-phenotype causal models in neuroscience, researchers remain mindful of the complications these many intricacies entail. Moreover, it is imperative that researchers be attentive to the details and possible sources of error contained in the various causal models that can be constructed before deciding which would be the most appropriate model to evaluate the data collected. This should be the case especially in studies employing deep phenotypes for neurobehavioral and neuropsychiatric disorders. Unlike experiments in which study environments are controlled, causes manipulated, and subsequent outcomes observed and measured, observational studies in which causal inferences are drawn post hoc from the data collected are prone to bias and false discoveries inherent in the enterprise. And those factors will most certainly affect the reliability and validity of the genotype-phenotype relationships constructed. Consequently, researchers should probably consider their chosen causal models as exploratory tools.
The authors declare no conflict of interest.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
Fisch GS. Deep Phenotyping in the -Omic Era: Methodological Issues Facing Neuropsychiatric Disorders. J Psychiatry Brain Sci. 2018; 3(6): 11; https://doi.org/10.20900/jpbs.20180011

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




