Location:Home >> Detail
J Psychiatry Brain Sci. 2017; 2(1): 1; https://doi.org/10.20900/jpbs.20170001

1 Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences and Yunnan Province, Kunming Institute of Zoology, Kunming, Yunnan, China
2 The Department of Nutrition, Harvard School of Public Health, Boston, MA, USA
3 Epidemiology Domain, Saw Swee Hock School of Public Health and Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
4 Department of Epidemiology, School of Public Health and Tropical Medicine, Tulane University, New Orleans, LA, USA
*Corresponding Author: Ming Li, Ph.D., Kunming Institute of Zoology, Chinese Academy of Sciences, 32 East Jiao-Chang Rd., Kunming, Yunnan 650223, China.
Background: Genome-wide scans have revealed that Darwinian natural selection affects human genome during human evolution, helping us to optimize complex human adaptations. Natural selection is also reflected in significant associations with human phenotypes, e.g., single genomic locus associated with diverse complex disorders and traits.
Method: Meta-analyses of single nucleotide polymorphisms (SNPs) in 10q24.33 with schizophrenia in Asian populations were conducted. Population genetic analyses of the schizophrenia risk SNPs were also conducted in diverse populations.
Results: We report a SNP rs11191580 in 10q24.33 to be significantly associated with schizophrenia in Asians. Further pleiotropic analyses reveal that the schizophrenia non-risk allele [C] at rs11191580 also reduces risk of blood pressure and hypertension in both European and Asian populations. The non-risk allele [C] is present in populations from Europe and Asia mainland, but totally absent in Africans. Further evolutionary analyses show that the derived allele (i.e., C-allele) has experienced recent positive selection in Europeans and Asians. Through subsequent exploratory analysis, we propose that the colder environment in Europe and Asia is likely the selection pressure, i.e., when modern humans migrated “out of Africa” and moved to Europe and Asia mainland (the continent colder than Africa), new allele derived due to positive selection. This new allele probably helped human beings to adapt to the cold environment and also protected them from risk of hypertension.
Conclusion: The current data provides an intriguing example demonstrating the reflection of natural selection on pleiotropic genomic locus, illustrating a possible mechanism of selection pressures in human populations.
Schizophrenia is a severe and common complex disorder with a lifetime prevalence of ~ 0.5 % among world populations. [1] Though heritability estimate of schizophrenia exceeds 80 %, [2] alleles identified to date explain only a small proportion of this rate. Until recently, large-scale genome-wide association studies (GWAS) have explained the polygenic architecture of schizophrenia, [3] i.e. a large number of single nucleotide polymorphisms (SNPs) having low to moderate effect sizes and are variable in statistical associations in small sample set altogether account for the disease risk.
In addition to the increasing number of reportedschizophrenia associated SNPs, epidemiological studies have also identified multiple traits and diseases strongly associated with this illness, including blood pressure, hypertension and cardiovascular diseases etc. [4, 5] The etiologic relationship between these phenotypes is yet to be explained. One plausible hypothesis is the genetic pleiotropy, [6] which is the association of individual genetic variation with ≥ 2 phenotypes. In fact, there are often overlapping traits among the abovementioned phenotypes defined through behavioral or clinical investigations, indicating potential shared genetic influences.
However, the underlying evolutionary mechanisms of these genetic pleiotropic phenomena remain unclear. Darwinian natural selection has played (or is still playing) important roles in complex human adaptations during evolution, resulting in presence or disappearance of specific allele that could help us adapt to external environment or survive through severe disorders. Among these genomic loci affected by natural selection, many of them can still be identified through association studies with human phenotypes such as microcephaly. [7, 8] Also, loci showing pleiotropic effects are recognized as potential signatures that Darwinian natural selection might have acted on this genomic region. [9, 10] For example, we previously reported genetic pleiotropic effects of a schizophrenia SNP rs13107325 in SLC39A8 on diverse human diseases and traits including schizophrenia, obesity and blood pressure, which were likely resulted from the concomitantly observed strong Darwinian positive selection on this genomic locus. [11]
Chromosome 10q24.33 is one of genomic regions most signi f icant ly associated with schizophrenia in recent GWAS among European populations, [3] but the associations in other ethnic groups are still unclear. Here, we have performed a series of convergent analyses on the evolutionary mechanisms underlying this genomic region. First, using a large-scale analysis in nine independent samples, we confirmed that the risk SNP rs11191580 in 10q24.33 was also significantly associated with schizophrenia in Asian samples, supporting the shared genetic risk of schizophrenia on this locus between continental populations. We then assessed the impact of the risk SNP rs11191580 on local gene expression in brain samples, and found that the SNP was associated with the expression of a humanspecific isoform of AS3MT. We also showed that rs11191580 in 10q24.33 region exhibited pleiotropic effects on diverse complex traits and diseases in both European and Asian populations, i.e., the schizophrenia non-risk allele [C] of rs11191580 was associated with decreased risk of blood pressure, hypertension and cardiovascular diseases. In addition, we demonstrated that the schizophrenia non-risk allele [C] of rs11191580 was present in both Europeans and Asians but absent in Africans, and the C-allele underwent recent positive selection in the populations from Europe and Asia mainland. This result indicated an origination of the allele after the modern human’s “out-of-Africa” scenario, and suggested that the selection pressure might be related to the local environmental factors in Europe and Asia, e.g., cold temperature of the local climate. This study further confirmed the theory that human evolutionary history acts on the changes of allele frequency of specific genomic loci and results in pleiotropic effects in complex human disorders/traits.
For the genetic analysis of rs11191580 with schizophrenia in Asian populations, seven casecontrol and two family-based samples were used, including a total of 13,042 cases, 18,435 controls and 1,115 nuclear families. All patients were diagnosed with schizophrenia according to either International Classification of Diseases 10 (ICD-10) or Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria, and control subjects were local volunteers with no record of mental illnesses.
Shaanxi, China. The Shaanxi sample included 1,430 schizophrenia patients and 1,570 controls. The subjects were Han Chinese recruited from First Affiliated Hospital of Xi'an Jiaotong University and Xi'an Mental Health Center. 53.4 % of the patients, were males (mean age 37.6 ± 11.2 years) and 46.6 % were females (mean age 35.1 ± 10.6 years). All patients were diagnosed by at least three psychiatrists according to the DSM-IV criterion. The control sample consisted of 54.8 % males (mean age 32.9 ± 10.9 years) and 45.2 % females (mean age 34.2 ± 11.7 years). Rs11191580 was genotyped with matrix-assisted laser desorption ionization–timeof- flight method by Guan et al. [12]
Yunnan, China. The Yunnan sample consisted of 547 unrelated schizophrenia patients (mean age 38.7 ± 10.6 years) and 755 unrelated healthy control subjects (mean age 36.8 ± 6.7 years). Rs11191580 was previously genotyped with SNaPShot method in this sample. [13] The patients were recruited from The Second People’s Hospital of Yuxi City and diagnosed as having schizophrenia by at least two psychiatrists according to the ICD-10 criterion. Potential participants who had the history of alcoholism, epilepsy, neurological diseases, or drug abuse were excluded. Meanwhile, unrelated healthy subjects were recruited from local community as control samples. These controls were all free of psychiatric disorders, drug abuse, alcohol dependence, or brain injury. All the patient and control subjects were Han Chinese.
Hunan, China. The Hunan sample contained a total of 976 unrelated schizophrenia patients (mean age 24.9 ± 8.3 years) and 1,043 matched healthy controls (mean age 37.4 ± 14.2 years). All subjects were Han Chinese recruited from Hunan Province in South Central China. In brief, patients were diagnosed independently by two experienced psychiatrists as schizophrenia according to DSM-IV criteria. Rs11191580 was genotyped with SNaPShot method in this sample by Ma et al. [14]
BIOX, China. The BIOX sample was comprised of two independent cohorts of 7,335 cases and 11,964 controls in total. [15] Participants in the schizophrenia group were from various mental health centers. The patients were interviewed by two independent psychiatrists and were diagnosed according to DSM-IV criteria. All healthy controls were randomly selected from Han Chinese volunteers who were asked to reply to a written invitation to evaluate their medical histories. The rs11191580 was not included in the original BIOX study, but a proxy SNP (rs732998, r2 = 1.0 with rs11191580 in Chinese) was genotyped, with Affymetrix Genome-Wide Human SNP Array and MassARRAY iPLEX Gold platform respectively.
Sichuan, China. The Sichuan sample was comprised of 1,087 schizophrenia cases and 1,063 healthy controls. All subjects recruited were of Han Chinese origin. Patients with schizophrenia were recruited from West China Hospital of Sichuan University in Sichuan, China and were assessed using the Structured Clinical Interview for DSM-IV Patient Version (SCID-P). Included patients fulfilled the diagnostic criteria for schizophrenia as specified in DSM-IV. The rs11191580 was genotyped with Illumina GoldenGate Assay in this sample. [16]
Guangxi, China. The Guangxi sample contained 462 schizophrenia patients and 598 healthy controls of Han Chinese origin as described previously. [17] The diagnosis of SCZ was confirmed independently by two experienced psychiatrists following the DSM-IV criteria. Rs11191580 was genotyped using Sequenom Mass-ARRAY® technology (Sequenom Inc., San Diego, CA, USA).
Asia family-based sample 01. This sample consisted of 536 affected families, comprising of 1,633 members including 698 schizophrenia patients. Rs11191580 was genotyped with TaqMan assay in this sample by Takahashi et al. [18] Diagnoses of all affected family members were based on either ICD-10 or DSM-IV criteria.
Asia family-based sample 02. This sample was comprised of 579 affected families of 2,296 individuals. Rs11191580 was genotyped in this sample using Illumina iSelect assay by Aberg et al. [19] Diagnoses of all affected family members were based on DSM-III-R diagnosis criteria.
Aichi, Japan. The Aichi sample included 1,032 unrelated schizophrenia patients (mean age 46.8 ± 14.8 years) and 993 unrelated healthy control subjects (mean age 49.7 ± 14.0 years). Rs11191580 was genotyped with Sequenom iPLEX Gold system (Sequenom, San Diego, CA) in this sample by Saito et al. [20] All the patients and control subjects were of Japanese origin. Patients were diagnosed as having schizophrenia according to the DSM-IV criteria.
Osaka, Japan. The Osaka sample was comprised of 173 schizophrenia patients (mean age 36.0 ± 12.3 years) and 449 healthy controls (mean age 35.4 ± 12.8 years). Rs11191580 was genotyped using TaqMan assay by Ohi et al. [21] All subjects were biologically unrelated within the second-degree of relationship and were of Japanese descent. Each patient with schizophrenia was diagnosed by at least two psychiatrists according to the criteria from DSMIV.
2.2 Statistical AnalysisWe used odds ratio (OR) and standard error (SE) from each individual cohort to estimate heterogeneity between different samples and to calculate the pooled OR and 95 % confidence interval (CI) in the combined samples.
In case-control studies, the allelic-specific OR and its associated SE of rs11191580 were calculated according to the number of allele accounts (or extracted from the studies) using RevMan 5.0.
In family-based studies, the OR of transmission disequilibrium test (TDT) and its associated SE were calculated as follows: [22]
OR=a/b
SE=SQRT(1/a+1/b)
Where “a” means number of times that A1 allele is transmitted from heterozygous parents to affected proband, and “b” means number of times that A1 allele is not transmitted.
To combine the results of individual samples, we calculated the heterogeneity between each study using the Cochran’s (Q) χ2 test, which is a weighted sum of the squares of the deviations of individual OR estimates from the overall estimate. In the absence of heterogeneity among individual studies, we used a fixed-effect model to combine the samples and to calculate the pooled OR and corresponding 95 % CIs; otherwise, a random-effect model was applied.
The meta-analysis was performed with the classical Inverse-Variance weighted methods using the R package (Meta module). We used a forest plot to graphically present the pooled ORs and the 95 % CIs. Each study was represented by a square in the plot, and the weight of each study was also shown.
2.3 MRI Imaging AnalysisWe recruited 294 unrelated healthy Han Chinese subjects (187 females and 107 males, mean age 35.1 ± 12.5 years). All volunteers were free of any history of mental disorders, drug abuse, alcohol dependence or brain injury. All individuals have provided written informed consents for participation. This imaging sample was previously shown to be effective for the detection of genetic effects on the features extracted from magnetic resonance imaging (MRI) data with negligible population stratification. [13,23] Structural MRI data were acquired using a Philips MRI scanner (Achieva Release 3.2.1.0) operating at 3 Tesla. High-resolution whole brain volumetric T1-weighted images were acquired sagittally with an inversion-recovery prepared 3-D spoiled gradient echo (SPGR) pulse sequence (Repetition Time = 7.38 ms, Echo Time = 3.42 ms, flip angle = 8, voxel dimensions = 1.04×1.04×1.80 mm3, slice thickness = 1.2 mm).
We used FSL-FIRST (FMRIB’s Integrated Registration and Segmentation Tool) from FMRIB Software Library (FSL) package to calculate the brain volume. [24] The shape/appearance models used in FIRST were constructed for 15 different subcortical structures using 336 manually segmented images. During registration, the input data (3D T1 images) were transformed to the MNI (Montreal Neurological Institute) 152 standard space by means of affine transformations based on 12 degrees of freedom. After subcortical registration, a sub-cortical mask was applied to locate the different subcortical structure. Segmentation based on shape models and voxel intensities was then conducted. All segmented subcortical regions were examined visually for registration or segmentation errors. Intracranial volume (ICV) as well as gray matter and white matter volume were calculated by removing non-brain material and segment into gray matter/ white matter/CSF using FSL-FAST. [25] Multiple linear regression analysis was performed to compare the brain volumes among different genotypes of rs11191580. Sex, age, age2, sex×age, sex×age2, and four principal components were included as covariates.
2.4 Expression Quantitative Trait Loci (eQTL) AnalysisFor eQTL analysis, we used the gene expression data from a recently published study[26] of 135 postnatal Caucasian non-psychiatric controls. In brief, the postmortem dorsolateral prefrontal cortex (DLPFC) samples were dissected and homogenized. 80−120M 100 bp paired end reads RNA-sequencing was then performed using Illumina Hi-Seq 2000 according to manufacturer’s protocol. Paired-end reads of cDNA sequences were aligned to the human genome reference (UCSC hg19) by spliceread mapper (TopHat v2.0.4), [27] providing known transcripts from Ensembl Build GRCh37.67. To quantify mRNA expression, we considered only the alignments in the 10q24.33 genomic region (corresponding to chr10:103605356–105615301 on genome build hg19). We controlled for expression heterogeneity due to potential latent factors using principal component analysis (PCA). Principal components (PCs) were calculated using the transformed (log base 2 with an offset of 1) read counts in chr10:103605356–105615301 region, and the first 10 PCs were used as covariates in the eQTL analyses. Genomic principal components were also calculated based on the genomic variants across common linkage disequilibrium independent variants across the genome, and the first five PCs were used as covariates in the eQTL analyses. Statistical analyses of mRNA expression associated with genotypes were conducted in R 3.0.1 using linear regression, treating log2-transformed expression levels as the outcome, and covaried for 10 expression PCs and five genomic PCs.
2.5 Analyses of Pleiotropic Effects of rs11191580 2.5.1 Coronary Artery DiseaseIn 2012, the CARDIoGRAMplusC4D Consortium performed an association analysis in 63,746 coronary artery disease (CAD) cases and 130,681 controls of European or Asian descent, [28] and identified 15 loci reaching genome-wide significance. The total number of susceptibility loci for CAD returned by this analysis was 46, and a further 104 independent variants were found to be strongly associated with CAD at a 5 % false discovery rate (FDR).
2.5.2 Blood Pressure and HypertensionThe International Consortium for Blood Pressure (ICBP) performed a multi-stage GWAS metaanalysis of blood pressure and hypertension in > 20,000 individuals of European ancestry. Phenotypes included in the analysis were systolic blood pressure (SBP), diastolic blood pressure (DBP), pulse pressure (PP, the difference between SBP and DBP), mean arterial pressure (MAP, a weighted average of SBP and DBP) and hypertension. [29, 30]
In another study, Kato et al. [31] conducted a metaanalysis of GWAS of SBP and DBP in >19,000 subjects of east Asian ancestry from the AGEN-BP consortium, followed by independent replications in east Asian samples.
2.5.3 Body Mass Index and ObesityObesity is a highly prevalent and heritable trait in global populations. [32] As a non-invasive and inexpensive measure of obesity, body mass index (BMI) has been extensively used to predict the risk of obesity-related complications. To better understand the biological basis of obesity, Speliotes et al. [33] conducted genetic association analyses between BMI and about 2.8 million SNPs in up to 123,865 individuals of European ancestry. They followed up 42 SNPs in up to 125,931 additional subjects and identified 18 new loci showing genomewide significant association with obesity. Detailed information about sample ascertainment and statistical analyses can be found in the original study [33] and GIANT website (http://www.broadinstitute.org/collaboration/giant/index.php/Main_Page).
Meanwhile, Wen et al. [34] conducted a metaanalysis of associations between BMI and ~ 2.5 million SNPs among 86,757 individuals of Asian ancestry, followed by in silico and de novo replication among 7,488−47,352 additional Asian-ancestry individuals, by which they identified several novel BMI-associated loci in Asians.
2.5.4 Energy IntakeDietary intake of macronutrients (carbohydrate, protein, and fat) is associated with risk of chronic conditions such as obesity and diabetes. [35] Two GWASs (discovery sample size N > 30 000 in each study) were conducted in independent samples of European descent to identify common genetic variants associated with macronutrients intake. [36, 37] The authors reported variants in FGF21 and FTO to exhibit genome-wide significant associations.
2.6 Samples for Population Genetic AnalysesWe used a total of 1,092 unrelated individuals from the 1000-Human-Genome Project, [38] including 379 Europeans (including 85 Utah residents with Northern and Western European ancestry; 88 Toscana from Italia; 14 Iberian from Spain; 93 Finnish from Finland; and 89 from England and Scotland), 286 Asians (consisted of 97 Han Chinese from Beijing, China; 100 Southern Han Chinese; and 89 Japanese from Tokyo, Japan), 181 Americans (comprised of 66 of Mexican ancestry from Los Angeles USA; 55 Puerto Ricans from Puerto Rico; and 60 Colombians from Medellin, Colombia) and 246 Africans (containing 88 Yoruba from Ibadan, Nigeria; 97 Luhya from Webuye, Kenya; 61 of African ancestry from Southwest USA), to analyze the frequency distribution of rs11191580 in different geographic populations, and to calculate the integrated haplotype score (iHS).
2.7 Tests of Natural SelectionThe iHS is a test typically designed to capture incomplete selective sweep for detection of recent positive selection. The iHS test statistic, which is calculated for common SNPs (minor allele frequency > 0.05) in the candidate region, reflects the differences in the long-range LD patterns containing the ancestral versus derived alleles. The genotype data was first phased, and then the iHS was calculated using software Selscan according to the methods implemented in previous studies. [39-42] SNPs under positive selection should have | iHS | > 2 (iHS < −2 means the derived allele undergoes positive selection and iHS > 2 indicates the ancestral allele undergoes positive selection), which corresponded to the most extreme 5 % of iHS values across the genome with minor allele frequency > 0.05.
In 2011, rs11191580 was reported to be significantly associated with schizophrenia in the GWAS by Psychiatric Genomics Consortium (PGC1) in populations of European descent (p = 1.11×10−8, OR=0.870), [43] and the significance became stronger in the later European GWAS meta-analysis (PGC1 + SWE) with an increased sample size (p = 2.09×10−10, OR = 0.827). [44] However, such associations were analyzed in Europeans, and the situations in other ethnic groups were rather limited with only one replication study in a smaller Han Chinese sample by a research group from Shaanxi, China (p = 3.03×10−4, OR = 0.808). [12]
Here, to further confirm if rs11191580 was associated with schizophrenia in general and larger Asian populations, we used additional data of rs11191580 from nine independent Asian samples with no overlap with the Shaanxi sample. Power analysis indicated that the present sample size showed a > 80% power of detecting significant association (p < 0.05). In these combined samples including 11,612 cases, 16,865 controls and 1,115 nuclear families, rs11191580 was significantly associated with schizophrenia (p = 1.99×10−3, OR = 0.942, 95 %CI = 0.907−0.978). When the Shaanxi sample was included in the meta-analysis, the association became stronger in a total of 13,042 cases, 18,435 controls and 1,115 families (p=4.78×10−5, OR = 0.928, 95 % CI = 0.895−0.962). There was no heterogeneity among these samples (I2 = 16.1 %, p= 0.295), and sensitivity analyses did not find changes of the significant association after removal of any single sample, implying that the association was reliable. The forest plot of meta-analysis in Asians is shown in Figure 1A. Although it did not achieve genome-wide level of statistical significance, the observed OR (0.928) of rs11191580 in Asian samples was in agreement with the European GWAS results on this SNP, suggesting that it was likely a real common risk SNP for schizophrenia in general populations.
 Figure 1 (A) Forest plot for meta-analysis of rs11191580 [C allele] with schizophrenia in Asian populations
Figure 1 (A) Forest plot for meta-analysis of rs11191580 [C allele] with schizophrenia in Asian populations
We then conducted the association analysis of rs11191580 with brain structures in an Asian sample of 294 healthy subjects. The results showed that rs11191580 was significantly associated with the total white matter volume (p = 0.019), and individuals carrying the non-risk allele (C) of rs11191580 tended to have larger white matter volumes (Figure 1B). These data implied that rs11191580 likely conferred risk of schizophrenia susceptibility through affecting brain development, especially formation/ development of white matter.
 Figure 1 (B) Association of rs11191580 with white matter volume in Asian populations.
Figure 1 (B) Association of rs11191580 with white matter volume in Asian populations.
In genetic association studies, an associated SNP most likely points to a larger region of correlated variants exhibiting high degree of linkage disequilibrium (LD), making it difficult to precisely identify the causal variant. To investigate if there were SNPs linked with rs11191580, we explored the LD between rs11191580 and surrounding SNPs. A proxy search for SNPs of LD with rs11191580 was performed on the SNAP website with European and Asian panels from the 1000-Human-Genomes (pilot 1) dataset. This search found that there were extensive SNPs in relative high LD (r2 > 0.80) with rs11191580, spanning a large genomic region covering CYP17A1, BORCS7, AS3MT, CNNM2 and NT5C2 (Figure S1). Thus we could not exclude the possibility that rs11191580 was only a marker reflecting the association signal due to its LD with the causative variant.
 Figure S1 A proxy search for SNPs of LD with rs11191580 in European and Asian samples from the
1000-Human-Genomes.
Figure S1 A proxy search for SNPs of LD with rs11191580 in European and Asian samples from the
1000-Human-Genomes.
The associations of rs11191580 with schizophrenia lent statistical support to the involvement of this genomic region in the risk of the disorder. However, these findings did not identify the underlying molecular mechanism. To test the effects of the risk SNPs on the expression of nearby genes, we used a RNA-seq sample in the dorsolateral prefrontal cortex (DLPFC) of European healthy controls (n = 135). As shown in Figure S1, rs11191580 and its high LD SNPs spanned CYP17A1, BORCS7, AS3MT, CNNM2 and NT5C2 in genome, therefore we focused on these five genes.
The expression quantitative trait loci (eQTL) analysis on the gene level found that rs11191580 was only associated with the expression of AS3MT but not the other four genes in this genomic region (p = 2.87×10−5, Figure 2A). A recent study suggested that there was alternative splicing within the AS3MT gene to result in two primary isoforms, one was the full length AS3MT (named AS3MTfull), and the other was a truncated isoform missing exon 2 and 3 compared with AS3MTfull (named AS3MTd2d3). [26] The transcript level analysis found that rs11191580 was only associated with the human unique isoform AS3MTd2d3 (p = 0.0095, Figure 2B). [26]
 Figure 2 (A) Association of rs11191580 with nearby gene expression.
Figure 2 (A) Association of rs11191580 with nearby gene expression.
 Figure 2 (B) Association of rs11191580 with AS3MTd2d3 and AS3MTfull isoform expression.
Figure 2 (B) Association of rs11191580 with AS3MTd2d3 and AS3MTfull isoform expression.
Accumulating lines of evidence have suggested substantial overlap of risk factors between schizophrenia and non-psychiatric disorders such as blood pressure, cardiovascular diseases (CAD) and hypertension from clinical, epidemiological and genetic perspectives. We then investigated if rs11191580 was also associated with these complex traits and diseases in both European and Asian populations. Interestingly, rs11191580 was significantly associated with blood pressure and CAD in both European and Asian populations (Table 1), and the schizophrenia non-risk [C] allele reduced risk of hypertension and CAD, which was in concordant with higher prevalence of hypertension and CAD in patients with schizophrenia across previous studies.
 Table 1. Pleiotropic effects of rs11191580 on complex diseases and traits
Table 1. Pleiotropic effects of rs11191580 on complex diseases and traits
Together with the results of eQTL analysis, these data suggested that the human-unique AS3MTd2d3 isoform might be also involved in the development of metabolic syndromes such as CAD and hypertension, but further studies are needed.
3.4 Evidence of Recent Positive Selection on rs11191580 in Europeans and AsiansPleiotropic effects of a genomic locus on diverse phenotypes might be a reflection of natural selection (such example can be referred to rs13107325 in SLC39A8 [11]). To test this hypothesis, we performed population genetic analysis of rs11191580 using the genotype data from 1000-Human-Genome.
The geographic distribution analysis found that the frequencies of the derived allele (i.e., schizophrenia non-risk C-allele) of rs11191580 were maintained at ~ 25 % in Asians and ~ 10 % in Europeans, but totally absent in Africans. These data indicated a clear regional distribution pattern and a relatively young origin of this SNP (Figure 1C).
 Figure 1 (C) Global distributions of rs11191580 in
world populations.
Figure 1 (C) Global distributions of rs11191580 in
world populations.
The regional enrichment of C-allele of rs11191580 in populations outside Africa mainland can be explained either by population substructure due to random genetic drift, or by recent positive selections in regional European and Asian populations. Longrange haplotype (LRH) tests can detect partial selective sweeps, particularly with allele frequencies as low as ~ 10 %. Therefore, we calculated the integrated haplotype score (iHS) (a statistic based on the extent of decay of LD surrounding a variant subjected to natural selection) of rs11191580 using the LRH tests. The iHS analysis of individuals from 1000-Human-Genome revealed that the derived C-allele experienced recent positive selection in Europeans (iHS = −2.16, p < 0.05), Asians (iHS = −2.22, p < 0.05) and Americans (iHS = −2.29, p < 0.05). In Africans, rs11191580 was monomorphic and had no iHS value. We also calculated the iHS of extensive common SNPs (N > 600) in chromosome 10, and found that rs11191580 was among those with the extremely 5 % lowest iHS values (Figure 3A−C). These data supported our hypothesis that rs11191580 underwent recent positive selection in European and Asian populations.

Figure 3. The distributions of iHS values for markers on Chromosome 10 in Europeans (A), Asians (B) and Americans (C)
The association between rs11191580 with diverse human phenotypes and its recent positive selection in both Europeans and Asians are intriguing. The derivation of the new allele reduces risk of both schizophrenia and cardiovascular diseases in Europeans and Asians. Considering that modern humans migrated out of Africa mainland about 40,000 years ago, [45] we speculated that this positive selection occurred after the arrival of modern human ancestors at Europe and Asia mainland given the evidence for a more recent positive selection event ( < 30,000 years old) revealed by the LRH tests. Also, since the positive selection acted on both European and Asian populations but not Africans, the selection pressure might be related to historical events shared by Europe and Asia but not Africa.
3.5.1 Hypothesized selection pressure related to obesity.To our knowledge, one of the biggest environmental changes when modern humans migrated out of Africa mainland was the decreased ambient temperature. Considering that Europe and Asia continents were much colder than Africa mainland, the new alleles (e.g., rs11191580 C-allele) probably arose when modern humans migrated to the changed environment to help promote energy intake and expenditure to maintain thermal homeostasis (i.e., an optimal body temperature that is most often above their ambient temperature) in the colder climate. However, the same alleles also contributed to the contemporary increase in obesity rate. Notably, this hypothesis is supported by the significant associations between rs11191580 with obesity and BMI in both Europeans and Asians (Table 2), with the positive selected allele (theschizophrenia non-risk C-allele) increased risk of obesity. The positive selected (C) allele is also associated with higher caloric intake from protein (Table 2), which is consistent with its role in obesity and further supports our speculation of the potential pressure of the positive selection on this genomic locus.
 Table 2. Effects of rs11191580 on obesity and energy intake
Table 2. Effects of rs11191580 on obesity and energy intake
It is known that when entering a new environment, e.g. another continent, some ancestral alleles which had been beneficial in Africa may become deleterious. [46] Under such conditions, some new alleles arose because of positive selection and protected humans from maladaptation to the new environment and development of diseases. [47] In this study, it is compelling to observe that the derived C-allele of rs11191580 reduces blood pressure and risk of hypertension in both Europeans and Asians. One of the most prevalent evolutionary hypotheses about hypertension (and blood pressure) is the “sodium hypothesis”, [46, 48] which argues that natural selection for sodium conservation in the hot, dry savannah climate (e.g., Africa) into which humans first emerged could have resulted in sodium avidity that today is maladaptive. As humans migrated out of Africa, selection pressure for sodium conservation was expected to decline in the cooler and wetter climates of the northern latitudes (e.g., Europe and Asia). As a result, ancestral sodium-conserving genotypes (the called “thrifty genotype”) were expected to persist as the result of genetic drift, and might lead to an increased risk of hypertension in a changed environment of sodium abundance (e.g., Europe and Asia) where they changed the set point of the renal pressure-natriuresis to a higher blood pressure range. In this case, the new alleles arose and underwent positive selection in the cooler and wetter climates (e.g., Europe and Asia), leading to reduced risk of hypertension. This hypothesis has been validated by previous observations on several hypertension candidate genes (e.g., CYP3A and AGT),[49, 50] and rs11191580 in 10q24.33 region is likely another example of such positive selection. Therefore, the protective effects of C-allele in rs11191580 from hypertension and blood pressures may reflect another pressure for this positive selection.
Taken together, we propose the intercorrelated relationship between positive selection of host alleles, hypertension, energy intake, and the human evolutionary histories (Figure 4). The positive selected risk C-allele might be beneficial for humans to better adapt to the Europe and Asia environment.
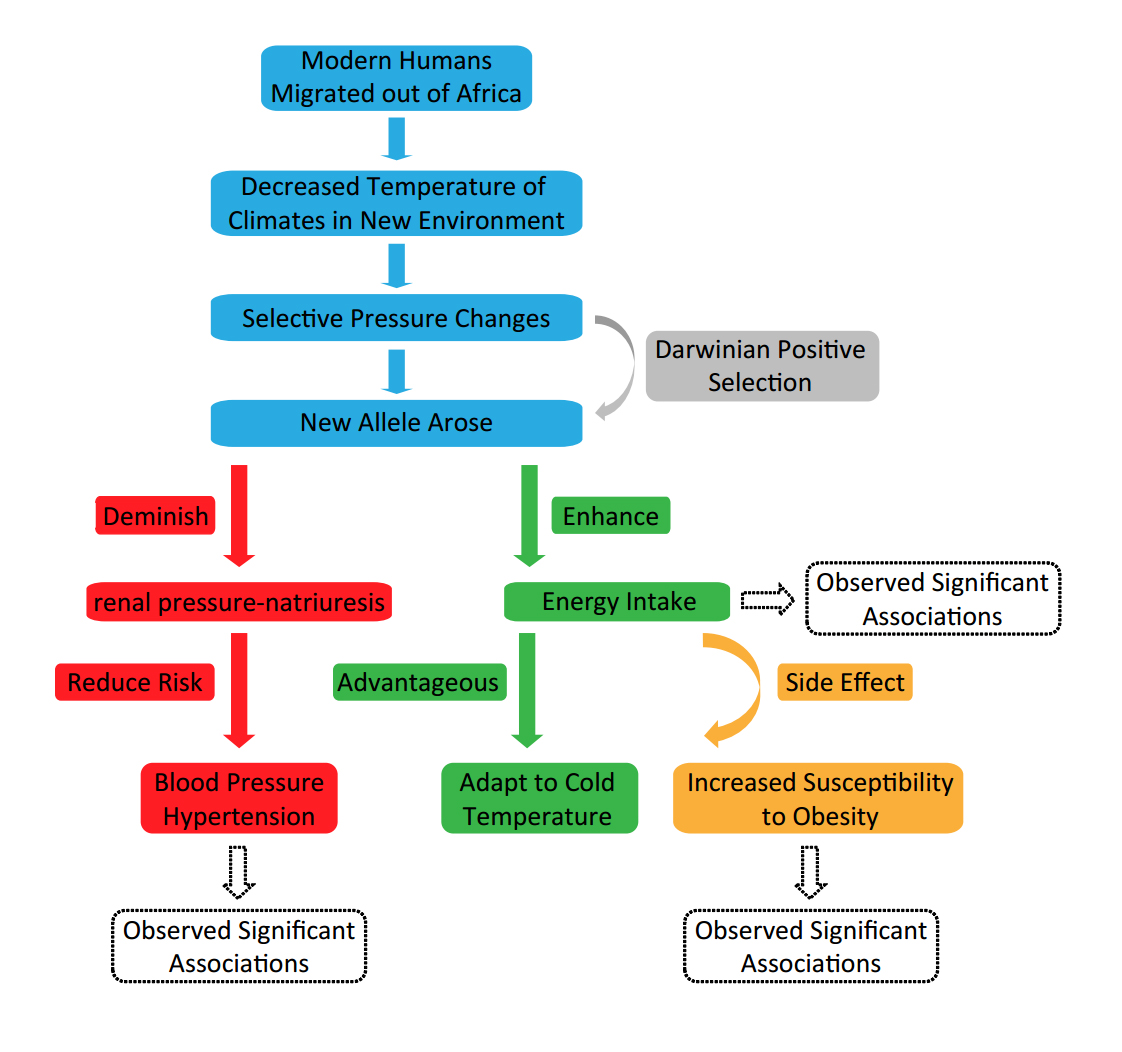 Figure 4. Hypotheses about pressures of positive selection on rs11191580 in European and Asian
populations.
Figure 4. Hypotheses about pressures of positive selection on rs11191580 in European and Asian
populations.
Chromosome 10q24.33 is one of the most significant loci for schizophrenia in recent GWAS among European populations, [3] but the associations in other ethnic groups are relatively unclear. Using independent samples from multiple Asian area, we confirm that rs11191580 in 10q24.33 is also associated with schizophrenia in this distinct population. Although it did not achieve the genomewide level of statistical significance, this observation was reasonable considering the limited sample size. We also show that the risk SNP is nominally associated with the brain structure variation, adding further evidence for the involvement of this genomic region in brain development and schizophrenia pathogenesis. We further confirm that rs11191580 is associated with the expression of a human-unique isoform of AS3MT, which is consistent with the previous study. [26]
Interestingly, the schizophrenia risk SNP is also associated with hypertension and CAD in both European and Asian populations. After analyses of this SNP with multiple metabolic malfunction related conditions, we observe the same direction of allelic effects on all conditions, suggesting pleiotropic effects of this genomic sequence on diverse human phenotypes. This may explain partially the observed comorbidity of schizophrenia and hypertension/ CAD. It is worth noting that the schizophrenia nonrisk C-allele at rs11191580 reduces risk of several metabolic syndromes (e.g., hypertension and CAD), which is also consistent with the higher prevalence of metabolic syndromes among schizophrenic individuals. [51] Moreover, the significant associations of rs11191580 with AS3MTd2d3 expression implies that AS3MTd2d3 may be a potential drug target for both metabolic syndromes and schizophrenia.
The pleiotropic effects on diverse human phenotypes lead us to perform the population genetic analysis on this genomic locus to understand its molecular evolution mechanisms. We have observed recent positive selection on rs11191580 that results in an increased frequency of the derived C-allele (schizophrenia non-risk allele) in Europeans and Asians. Considering that modern humans migrated out of Africa mainland about 40,000 years ago, we speculate that this positive selection occurred after the arrival of modern human ancestors at Europe and Asia continents. Since the long-range haplotype (LRH) tests find evidence for a more recent positive selection event (< 30,000 years old), the pressure of this selection may be related to common historical events in Europe and Asia but not Africa, such as the colder climate. However, cautions should also be taken in interpreting these results, i.e., although we identified significant associations of rs11191580 with multiple phenotypes in distinct populations as well as the signature of recent positive selection on this locus, whether rs11191580 is the causative variant remains inconclusive, and the positive selection may act on a large genomic region due to the observed high LD in both populations. To nail down the exact risk variant, further studies are necessary.
Given the clear positive selection on this genomic locus, we propose two possible selection pressures. The first one is related to energy intake and its byproduct obesity, while the other one is associated with blood pressure and hypertension. However, we do not identify selection pressure related to schizophrenia as the newly derived allele of rs11191580 confers reduced risk of schizophrenia in both populations. This phenomenon suggests that Darwinian positive selection may also protect humans from schizophrenia, but the relevant mechanism awaits further investigation. Also, it should be noted that the lifetime prevalence of schizophrenia does not differ significantly between continental populations. Since schizophrenia is a complex disorder involving thousands of alleles (may be different sets of risk alleles between populations) with limited effect of each, the combined contributions of all these alleles probably result in the similar prevalence of the disorders worldwide. We thus presume that positive selection on schizophrenia alleles would not be restricted within single populations, and there should be other risk alleles under positive selection in different populations (such as African), which are yet to be identified.
In summary, we have identified a SNP rs11191580 showing pleiotropic effects with diverse human phenotypes in Europeans and Asians, and we detected evidence of positive selection on the derived allele in both populations. Our evolutionary and genetic analyses may offer a unique and powerful opportunity to bring approaches from different perspectives together to discover how and why human diseases risks have evolved. [52]
The authors declare no conflict of interest.
This work was supported by CAS Pioneer Hundred Talents Program (to M.L.). The authors are deeply grateful for Miaoxin Li and Pak-Chung Sham (The University of Hongkong, China) for sharing their results of Sichuan samples. The authors also thank CHARGE Nutrition Working Group and DietGen Consortium for sharing their results about energy intake.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
Li L, Chang H, Huang T, Zheng Y, Qi L, Xiao X, Li M. Recent Positive Selection Drives the Expansion of a Schizophrenia-associated Variant within 10q24.33 in Human Populations through Its Pleiotropic Effects on Diverse Human Complex Traits. J Psychiatry Brain Sci. 2017; 2(1): 1; https://doi.org/10.20900/jpbs.20170001

Copyright © 2020 Hapres Co., Ltd. Privacy Policy | Terms and Conditions




